Mi mindent mond el a történetünkről egyetlen sejtünk? Szöllősi Gergely Az atomoktól a csillagokig című sorozat keretén belül a biológiában használatos fizikai modellekről adott elő: a kutatók ugyanis olyan modelleket dolgoztak ki, amelyekkel a génfákat bele tudjuk rajzolni a fajfákba, a kapott eredmények pedig átfednek a fosszíliákkal. Olvassátok el Püski Bianka tudósítását, ha meg akarjátok tudni, hogy „mit mondanak a genomok az evolúcióról, és hogyan?”
Szöllősi Gergely, az ELTE Biológiai Fizika Tanszék ERC-díjas és Lendület-nyertes kutatója tartott előadást március 25-én a molekuláris evolúció aranykoráról, azaz az általa „biológia 2.0-nak” aposztrofált időszakról Az atomoktól a csillagokig című ismeretterjesztő előadássorozat keretein belül, élőben, a Galileo Webcast közvetítésében. Bizonyára felmerül benned is a kérdés, hogy az evolúcióhoz vagy éppen a molekuláris genetikához – amelyek alapvetően biológiai és biokémiai témák – mégis mi köze lehet egy fizikusnak? Ígérem, a tudósításból erre és még sok érdekes kérdésre választ fogsz kapni, mint például, hogy hogyan képes akár a te egyetlen parányi sejted is mesélni a földi élet történetéről! Az előadást pedig vissza tudod nézni ezen a linken.
Szöllősi Gergely Debrecenben végezte el a gimnáziumot fizika tagozaton. Már egészen korán körvonalazódott benne az élet nagy, „Mi leszek, ha nagy leszek?” kérdésére a saját, egészen specifikus válasza: elméleti fizikus. A pályaválasztás kapcsán egyértelmű volt, hogy céljainak megvalósításához a legjobb hely az általa „a magyar fizika fellegvárának” aposztrofált ELTE Fizika Tanszéke, ahol a doktori képzést is végezte. Pályaválasztása, illetve pályája kapcsán meghatározó időszakot jelentett a Lyonban töltött három év, amely során a genomika, azaz a szervezetek teljes genomjának vizsgálatával foglalkozó tudomány területén gomba módjára szaporodó szekvenciális adatok rendszerezéséről, illetve az értelmezésükhöz használatos modellek kidolgozásáról szóló kutatások részese lehetett. Jelen előadásában többek között a lyoni tapasztalatait osztotta meg velünk, mely időszakot követően vissza is tért Budapestre, s ma is aktív kutatómunkát végez az ELTE Fizikai Intézetének keretein belül. De miért foglalkozik egy fizikus az evolúcióval?
Az előadás felépítését követve elengedhetetlen néhány, a témában releváns fogalom, valamint azon hipotézisek ismertetése, amelyek bár megdőltek, de kétségkívül elengedhetetlen lépcsőfoknak bizonyultak a tudomány jelenlegi szintjének eléréséhez. Itt meg is ragadnám az alkalmat egy nagyon fontos gondolat megjegyzésére: a tudományban pont az a szép, hogy önellenőrző, a saját, esetlegesen hibás következtetéseiből tanul, fejlődik. Ezért, ha találkozol egy tudományos állítással – a koronavírus kapcsán ez különösen aktuális élmény – , amelynek egy hét múlva az ellenkezőjét állító álláspont van érvényben, ne rendüljön meg a tudományba, illetve annak művelőibe vetett hited. Sőt, pont ellenkezőleg, inkább örülni is lehet annak, hogy ennyire rövid idő elég ahhoz, hogy egy nem teljesen megalapozott vagy helytelen következtetés javításra kerüljön.
Szöllősi Gergely már az előadása kezdetén leszögezte (talán nem is teljesen tudatosan), amikor kutatási területének megválasztását indokolta, hogy az evolúció, illetve maga a genomika is multidiszciplináris területek. A kutató számára a biológia tartogatja a legizgalmasabb kérdéseket: „a legtöbb nyitott kérdés a biológiában van”. Ahogy elmondta: „A molekuláris evolúciókutatás aranykora zajlik”, ezt a jelenséget a The Economist folyóirattól kölcsönzött biológia 2.0 kifejezéssel nevezte meg a kutató. Számos fontos tudományos áttörésre, elméletre volt szükség ahhoz, hogy a tudomány eljusson a mai fejlettségi szintjére, de nézzük végig, hogy a témában mely állomások voltak a legfontosabbak! 1953 („Sztálin halála mellett”, jegyezte meg humorosan Szöllősi Gergely) a biológia történetében is egy történelmi év, ugyanis Watson és Crick felfedezték, illetve modellezték a DNS-t.
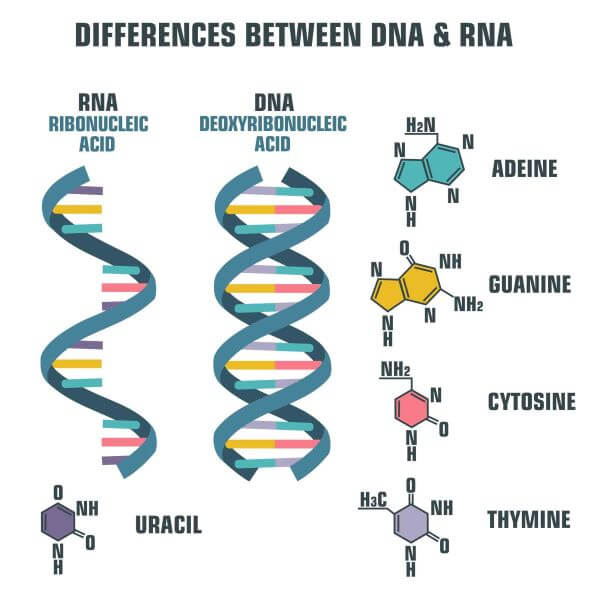
A DNS kettős hélix, valamint a DNS-t felépítő nukleotidok, azaz a bázisok szerkezete. A kép forrása: https://biologydictionary.net/dna-vs-rna/
Mi is az a DNS? Röviden megválaszolom a költőinek ható kérdésem, hogy az előadásban elhangzó ismeretek érthetőek legyenek. Most biztosan sokan magukban hadarják, hogy dezoxiribonukleinsav, de mit is jelent ez valójában? Komplementerként kapcsolódó (C-G, A-T) bázispárokat, a molekula a genetikai öröklés szubsztrátuma. Fontos, hogy a bázisok szigorúan C-G, azaz citozin-guanin és A-T, azaz adenin-timin felállásban állhatnak párba. Természetesen ennek molekuláris okai vannak, de most elég, ha megjegyezzük, hogy a bázis óvodában a bázisoknak állandó társa van, akinek mindig meg kell fogniuk a kezét, amikor az óvodából fagyizni, vagy a fogászatra indulnak. A bázisok párosodása teszi lehetővé magának a genetikai információnak a másolhatóságát, reprodukálhatóságát, azaz magát az öröklődést is, ami könnyen beláthatóan magának az evolúciónak is az alapja. Adott tehát a DNS kettős hélix, amit elképzelhetünk úgy is, mint egy összecipzározott pulóvert, ahol a cipzár két szárában lévő fogacskák a bázisok. A reprodukálhatóság érdekében a DNS kettős hélix kitekeredik, azaz kicipzározódik a pulóver, egy DNS-polimeráz nevű enzim pedig bázisról bázisra haladva „kiegészíti”, vagy ha úgy tetszik, tükrözve lemásolja a hélix egyik szálát. Voilà! Kész a DNS-lánc másolata!
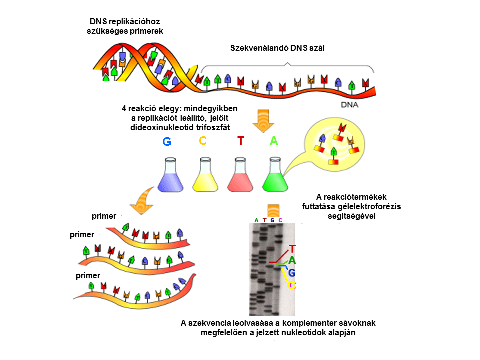
A Sanger-féle szekvenálás módszerének lépései. A kép forrása: Népegészségügyi genomika | Digitális Tankönyvtár (tankonyvtar.hu)
Tehát már tudjuk, hogy milyen a szerkezete a DNS-nek, ismerjük, hogy hogyan működik a reprodukciója, de mégis hogyan lehetne szekvenálni, előállítani? Bár Maxam és Gilbert dolgozta ki magának a szekvenálásnak a kémiai módszerét, de azt a gyakorlati eljárást, amit ma is használnak, illetve a mai napig a szekvenálás alapját képezi, azt 1977-ben Frederick Sanger dolgozta ki. Először a szekvenálni kívánt DNS-szálat denaturálta, majd a leolvasni kívánt szálhoz (emlékezzünk a cipzáras hasonlatra) egy komplementer, rövid szálat párosított, ez a primer. Ehhez öntött dezoxi, azaz fogalmazzunk úgy, hogy rendes nukleotidokat és nem rendes, úgynevezett didezoxinukleotidokat, amiket fluoreszcens festékkel, különböző színekkel jelölt. Amennyiben nem rendes, azaz didezoxinukleotidok épülnek be a szintetizálódó új szálba, a reakció leáll. A szekvenálási reakciók során eltérő hosszúságúak lesznek a láncok, illetve mindegyik láncnak fluoreszcensen jelölt végei lesznek, amik más és más hullámhosszon fognak fluoreszkálni, azaz más színűek lesznek attól függően, hogy melyik nem rendes nukleotid épült be a láncba. A színek alapján pedig megtudjuk, hogy a lánc milyen bázisra végződik. Mivel minden jel egy bázissal hosszabb, mint az előző, ezért tulajdonképpen magát a DNS-szálnak a bázissorendjét tudjuk leolvasni. Hatalmas előrelépést jelentett az evolúciós kutatásokban is a DNS szekvenálásának sikeres módszertana, hiszen a kétezres éveket meghatározó Humán Genom Projekt számos meglepetést okozott rendszertani szempontból, arról nem is beszélve, hogy robbanásszerűen megnőtt a rendelkezésre álló adatok száma, miközben időben napjainkhoz közeledve exponenciálisan csökkent a szekvenálás költsége. A számok embereinek: 2002-ben 1000 dollárba került 1 milliárd bázispár szekvenálása, addig napjainkban ez centekben mérhető összeg. A technikai fejlődés persze rohamos, gondoljunk csak bele, hogy 1997-ben milyen nagy szenzáció volt, hogy a Deep Blue megverte Garri Kaszparovot, az „emberi” sakkozás akkori csúcstartó világbajnokát, napjainkban pedig mindenki olyan mobiltelefonokkal a zsebében szaladgál, amelyekre legalább tízféle sakkszimulációt tud letölteni. Adott tehát az adatok forradalma a biológia területén is, ami nem meglepő, hiszen lassan le kell cserélnünk a „színház az egész világ” szállóigéjét „adat az egész világ”-ra. De hogyan lehet a rendelkezésünkre álló hatalmas mennyiségű adatot statisztikus, matematikai modellek révén értelmezni, segítségükkel evolúciós történeteket rekonstruálni? Itt jön képbe a fizikus.
Önmagában az élővilág sokfélesége is rengeteg adat, és közel sem ismerjük mindet, sőt! Gondoljunk csak bele, hogy a tudósok, kutatók körülbelül 1,7 millió növény- és állatfajt azonosítottak és neveztek el, míg a legoptimistább biológusi becslések szerint is minimum 10-15 millió faj él napjainkban a Földön. Természetesen az biztosabb, hogy nagytestű emlősök nem nagyon maradnak felfedezetlenül, de Dél-Afrikában a 3-4 ezer leírt, rendszerezett pókfaj száma körülbelül csak a fele lehet annak, ahány faj valójában előfordulhat a területen. Akkor mi a helyzet a genomokkal? Egyáltalán mi a fene az a genom? Miért szolgáltat informatív adatokat evolúciós szempontból?
A (humán) genom tulajdonképpen a „genetikai tervrajzunk, mind a 100.000 milliárd sejtünkben”. Szemléletes példa lehet a tervrajz mivoltára az első állati klónozás. A bárány klónozása során genetikailag teljesen megegyező egyedet „hoztak létre”, amihez elég volt egy nem megtermékenyített, de érett petesejt magját eltávolítani, s annak a helyére a klónozni kívánt egyed egy emlőmirigyének sejtmagját beültetni, majd a módosított petesejtet egy „béranya” bárányba ültetni, ahol az egyed DNS-éből megszületett a pontos másolat, azaz Dolly. Tehát Dolly tökéletesen reprodukálható volt az eredeti egyed DNS-ében tárolt információból. A genom tehát olyan, mint az IKEA-s bútorok, megegyező tervrajzok alapján végtelen számú ugyanolyan bútor konstruálható, persze szerencsére a mi tervrajzaink egyénenként különböznek. A genomba tartoznak tehát maguk a gének, azaz a nukleinsavak a DNS-szakaszokban, amelyek a fehérjék előállításához szükséges információt tartalmazzák, az úgynevezett nem kódoló szakaszokkal együtt, azaz a saját genomunk a kromoszómakészletünk teljes DNS-szekvenciáját jelenti. Mit jelent ez számokban?
A Humán Genom Projekt kétségkívül a 2000-es évek egyik legmeghatározóbb tudományos projektje, áttörése. 1990-től 2006-ig rengeteg nemzetközi tudós involválódott a projektbe, hogy a teljes emberi genomot feltárják a nukleotidok, azaz a bázisok sorrendjének szintjéig. Az ember 23 kromoszómája esetén ez körülbelül 3 milliárd bázispárt jelent, ami 6 GB-nyi adatot és körülbelül 30 000 gént jelent. Számos út nyílt meg a tudomány előtt ennek a nem elhanyagolható mennyiségű információnak a feltárása, valamint az egyre olcsóbbá váló szekvenálás révén. Jelentős előrelépést jelent például a génklónozás. Génklónozás történik például a PCR során is: miután például a jelenleg is zajló tömeges tesztelésekkor mintát vesznek az adott páciens orrnyálkahártyájáról, egy oldatba keverik, ahol a kémcsőben lévő enzim elindít egy úgynevezett polimeráz láncreakciót. Ezután néhány óra alatt az enzimek lemásolják a DNS-t, amelynek mennyisége lépésenként megkétszereződik, így a PCR alkalmassá válik tehát vírusok, mikrobák kimutatására; de például az egyiptomi múmiák, vagy több ezer éve halott állatok DNS-ének megsokszorozására is. Az ilyen típusú munkák rengeteget mesélhetnek az állatok és növények DNS-ében lévő génekről, amelyek megőrződtek a múltból, így például olyan elsődlegesen valaminek hangzó célok valósíthatóak meg, mint több ezer éve kihalt növények által termelt hatóanyagok sikeres szintézise és alkalmazása gyógyászati célokra.

Kép forrása: Make A Candy DNA Model | STEM Activity (sciencebuddies.org)
De mennyi információ van a genomban a múltunkról, illetve az élet több milliárd éves evolúciójáról? Erre a kérdésre keresi a választ Szöllősi Gergely. Fontos szétválasztani az aszexuális és a szexuális fajokat. Aszexuális fajok például a kettéosztódással szaporodó baktériumok. Az adott baktériumból létrejövő két utód egy DNS-replikációs eseményre vezethető vissza, azaz teljesen ugyanolyan a genetikai állományuk az utódoknak, nem különböztethetőek meg. Ezzel szemben a szexuálisan szaporodó fajok, mint például az ember esetében az ivarsejtek kialakulásához egy specifikusabb osztódás, a meiózis vezet, amelynek végeredménye, hogy egy sejtből négy ivarsejt keletkezik, és az osztódás során lezajló úgynevezett crossing over (átkereszteződések, kicserélődések) révén biztosított a genetikai változatosság. Ez megmagyarázza, hogy miért különbözünk és hasonlítunk egyszerre a testvérünkre. Ugyanazok a szüleink, azaz azonos genomkészletből indultunk ki, csak a meiózis során a tényleges létezésünkhöz szükséges ivarsejtek kialakulása során, majd a megtermékenyítést követően teljesen véletlenszerűen kaptunk 50-50%-ot a szüleinktől, ezért vannak tulajdonságok, amelyekben mind egymásra, mind szüleinkre hasonlítunk, és vannak tulajdonságok, melyekben inkább a nagypapára, vagy éppen egyik élő rokonunkra sem.
Korábban annak becslésére, hogy adott szekvenciáknak mikor élhetett a legkorábbi közös őse, a molekuláris óra elméletét használták, ezért érdemes megismernünk, annak ellenére, hogy mára inkább hipotézisként aposztrofálható. Ez a hipotézis feltételezi, hogy minden fajnak azonos a molekuláris ideje, azaz ugyanolyan gyorsan fejlődik, vagy ugyanolyan idős a sejtvonala; valamint azt vette alapul, hogy a DNS-másolás (replikáció) egészen pontos, de azért időnként előfordulnak benne hibák, amelyek mutációkat eredményeznek. Egy-egy mutáció pedig evolúciós változásokhoz vezet. Ezek a változások természetesen lehetnek előnyösek, de sajnos életképtelenséget is okozhatnak, ha mondjuk olyan helyen keletkezik a hiba, ami a fehérjeszintézis kezdetét jelöli ki. A molekuláris óra hipotézisét/elméletét követő tudósok nem csináltak mást, mint megszámolták a két összehasonlított szekvencia közötti eltérések számát, majd ezt számszerűsítették. Például Pauling és Zuckerkandl a ló és az ember hemoglobinjának szekvenciáját hasonlította össze, amelyek között 18 különbséget találtak, így a két faj kettéválását 130 millió évvel ezelőttre tették. Sajnos a helyzet természetesen nem ilyen egyszerű, a mutációk, a szekvenciák közötti különbségek nem adják meg az evolúciós időt, másrészről egyedi molekuláris órák jellemzik az egyes fajokat.
Egyelőre a fosszíliák geológiai rétegei alapján meghatározható kor az abszolút, de van lineáris összefüggés a molekuláris óra és a fosszíliák szerint megadható kor között. A valaha élt élőlények, evolúciós események időpontjait tehát a két módszer kombinálásával tudjuk megbecsülni.

A Mitokondriális Éva csak metaforikus elnevezés, önmagában nem bizonyítja a teremtéstörténetet. A kép forrása: https://hu.m.wikipedia.org/wiki/Mitokondriális_Éva
Mitokondriális Éva, azaz a legutóbbi közös ősünk körülbelül 100-150 ezer éve élhetett, ez a mai mitokondriális genom, illetve az „első” mitokondriális genom szekvenciái közti különbségből becsülhető. A mitokondriumaink tulajdonképpen a sejtjeink generátorai, energiaraktárai, amelyek maguk is evolúciós „kordokumentumok”. Ugyanis az endoszimbionta-elmélet értelmében egy prokarióta bekebelezése révén alakult ki, azaz valaha a sejtjeinkben lévő mitokondrium is prokarióta volt. Ezt bizonyítja a cirkuláris, saját DNS-e, saját genomja, ami 16,5 ezer bázispárból áll. Az Éva megnevezés pedig a teremtésmítosz szerinti (ős)anyának, Évának a metaforája, hiszen a mitokondriumunkat az anyukánktól kapjuk.
Miről mesél akár egyetlen darab parányi sejtünk? Következtethetünk például arra, hogy időben mekkora volt a populáció mérete. Megállapíthatunk például egy olyan összefüggést, hogy ha erősebb a szűkülés, azaz egy populáció mérete drasztikusan csökken, akkor ezzel párhuzamosan több is a közös ősökre visszavezethető esemény, hiszen ha kicsi a populáció, sokkal könnyebben találunk közös őst. Gondoljunk csak a teremtésmítosz allegóriájánál maradva arra, hogy a populáció egyetlen emberpárból áll; ebben az esetben minden utód rájuk vezethető vissza. Ahhoz, hogy pontosan meghatározható legyen a múltban élő populációk mérete, ismernünk kell a rekombinációs töréspontokat, illetve azokat a rokon szegmenseket, szekvenciákat, amelyek összehasonlítása informatív lehet. Sajnos egyiket sem ismerjük pontosan, ezért fontosak az úgynevezett szimulációk, melyek során ábrázolják az evolúciós folyamatokat, tehát például a mutációs eseményeket is.
Jogosan merülhet fel a kérdés, hogy ha az élővilág diverzitásának nagy részét nem ismerjük (ez a mikrobiális sötétanyag), ahogy nem ismerjük a rekombinációs töréspontokat sem, és csak szimulálni tudunk, akkor mégis miért aranykora ez a biológiának? Hát azért, mert rengeteg a megválaszolandó, nyitott kérdés. De akkor mi az, amit tudunk? Például evolúciós családfát rekonstruálni. Ehhez szükséges a rokon szekvenciák, szegmensek összevetése, a mutációs folyamatok modellezése, a változások szekvenciában való megjelenésének és megmaradásának folyamatainak ismerete. Részleteiben jelenleg ezt nem tudjuk modellezni, azonban azt, hogy milyen valószínűséggel fordulhat elő változás, azt az empirikus szubsztitúciós csere modell alkalmazásával, valószínűségszámítási, mátrix-exponencializálási képességekkel megoldható, „mindössze” azt kell számszerűsíteni, hogy mekkora a valószínűsége bizonyos szekvenciasorrend-előfordulásnak, -cserélődésnek. Például milyen valószínűséggel cserélődik A bázis T-re, vagy G bázis C-re? Természetesen vannak gyakoribb események, amelyek tapasztalati szempontból is beláthatók: például gyakrabban lesz A-ból G, mert mindkettő purin (két gyűrűs) molekula. A nukleotidok (bázisok) gyakorisága mellett tehát a szimmetrikus kicserélődési utakat is felhasználjuk, majd ez után tehetjük fel a legfontosabb kérdést: milyen génfa, illetve evolúciós modell generálta az adott szekvenciát?
Összességében megállapítható, hogy számos, statisztikai szempontból hasonló génfa van, azaz az egyes géntörténetek nagyon elmosódottak, arról nem is beszélve, hogy minket a fajfa és nem a génfa érdekel. Vagyis nem az egyes DNS-ek replikációs eseményei érdekelnek minket, hanem azok az események, amikor egy-egy faj kettéválik. Mi lehet a megoldás az elképesztően sok és változékony adat kiküszöbölésére?

Kép forrása: Molekuláris sejtbiológia | Digitális Tankönyvtár (tankonyvtar.hu)
Keresnünk kell egy olyan szekvenciát, amelyre kevésbé jellemző duplikáció és a diverzifikálódás, evolúciós értelemben konzervatív. Alkalmas lehet erre a riboszóma, illetve a riboszómális RNS (rRNS), aminek a fehérjeszintézisben van elengedhetetlenül fontos szerepe. Három bázisból aminosavat, a fehérjék alegységeit, építőköveit hozza ugyanis létre. Amennyiben ebben a folyamatban akár csak egy apró kis deficit is van, az utód életképtelen lesz, így a riboszóma, illetve az rRNS minden utódban ott van, lassan és konzervatívan változik: tehát alkalmas a fajfa rekonstruálásához. Ez azt jelenti, hogy génállományunk 1%-át használjuk az élet történetének megfejtéséhez. És ha ez nem lenne elég, az rRNS molekuláris hőmérőként is funkcionál: például ha magasabb egy élőlény rRNS szekvenciájában a G-C bázisok aránya, az adott élőlény optimális növekedési hőmérséklete magasabb (mint például a képen látható extremofil Thermus aquaticusnak), a hideghez alkalmazkodott élőlények esetében pedig jóval alacsonyabb az említett bázisok aránya.

Az extremofil Thermus aquaticus. Kép forrása: Coronavirus | El hallazgo en los manantiales de Yellowstone que fue clave para hacer los test de COVID-19 | TECNOLOGIA | EL COMERCIO PERÚ

A képen Bastien Bousseau disszertációjához készített festményt láthatunk, amelyet Bastien édesanyja készített. A kép forrása: Szöllősi Gergely előadásának slide-ja
Azt hitted, hogy ez lesz az a téma és előadás, ahol fikarcnyi bölcsészettel sem találkozol? Hát nem! Az előadó, illetve Bastien Bousseau doktori disszertációjában az evolúciókutatók helyzetét a platóni barlang-metafora analógiáján próbálta magyarázni. „A génfa a fajfa leképezése, a genomevolúció prizmáján keresztül” – állítja Szöllősi Gergely, azaz maga a kutató csak a szekvenciákat látja, amikből génfák rekonstruálhatóak, a fajfa a kutató számára láthatatlan marad. Az előbb említett szubsztitúciós modellt, ahol az egyes bázisok jelenlétének, illetve cserélődésének valószínűségét vettük alapul, kombinálnunk kell a génszületésekkel és -halálokkal, duplikációs eseményekkel; ugyanis rengeteg duplikációs esemény zajlott és zajlik is, de nem mind vezet fajkeletkezéshez. Az úgynevezett hierarchikus generatív modellben ötvözzük tehát a gének születésének és halálának valószínűségét, szorozva a szubsztitúciós paraméterekkel, ezáltal tehetünk közös statisztikai becslést a múltra.
Eddig láthattuk, hogy a rekombinációs mutációs események következtében a génfa nem mindig követi a fajfát. Ennek hátterében még egy fontos, eddig nem említett ok is áll, méghozzá a horizontális géntranszfer jelensége, azaz hogy (például egysejtűek esetében) kívülről is kerülhetnek gének a genomba, például vírusok által. Érdekes példát említett az előadó: a karotingyártó géneket. Tudjuk, hogy a növények, egyes algák termelik a karotint, míg az állatok vagy az ember elfogyasztják azt. 2010-ben azonban érdekes felfedezésre jutottak a kutatók a borsón élő levéltetvekkel kapcsolatban, megszekvenálták ugyanis a karotingyártó génjét, majd egy úgynevett „genom Google”-ben lefuttatva a keresést azt az eredményt kapták, hogy bizonyos gombák karotinszintéziséhez hasonlít. Azaz az állatok a LUCA-tól (Last Universal Common Ancestor, utolsó egyetemes közös ős) távolodva elvesztették a karotingyártó génjüket, de a gombák és növények ágán ez tovább öröklődött. Ezután egy szimbiózis kapcsán a levéltetű ősének genomjába bekerült a karotinszintézis gén, ami (mivel hasznos a ragadozók ellen) azóta is a genomjában maradt.

Karotingyártó génnel rendelkező levéltetű. A kép forrása: https://hu2.farmlux.biz/perec/vyrashchivanie/prichiny-poyavleniya-tli/
Sok kutató zajnak tartja a horizontális géntranszfereket az evolúciós életfa rekonstruálásával kapcsolatban, hiszen végtelen számú vízszintes kapcsolattal növelik az egyébként is végeláthatatlan, számos evolúciós kapcsolatot az egyes fajok között. Az előadó optimista, a transzferekre plusz információforrásként tekint. Ahogy mondta: „A transzferek tulajdonképpen molekuláris fosszíliák, amelyek a génszekvenciák időrendiségét tudják rögzíteni, kiegészítő információforrást adnak az időbeliségről.”
Összességében tehát megállapítható, hogy a rendszertanban és az evolúciókutatásban zajló reformkornak, illetve aranykornak koránt sincs vége, sőt, igazából szinte csak most kezdődik. Abszolút időről információt csak a fosszíliák hordoznak, de számos élőlényről akár a szerkezete miatt, akár több milliárd éves kora ellenére nem található fosszília, így önmagában kevés információt szolgáltatnak evolúciós léptékben. Ezért szükséges olyan empirikus modelleket kidolgozni, mint a Szöllősi Gergelyék által használt mátrix-modellek, vagy éppen a gének születésének-halálának valószínűségét számoló statisztikák. A transzferek pedig molekuláris fosszíliák, amelyek szekvenciákból rekonstruálhatóak; ezek a relatív korról adnak információt, hiszen a donor őseinek öregebbnek kell lennie, mint a recipiensnek.
Olyan modelleket dolgoztak tehát ki, amelyeken a génfákat bele tudjuk rajzolni a fajfákba, a kapott eredmények pedig átfednek a fosszíliákkal. Amit igazából mérni tudunk: az evolúciós ráta és az idő szorzata. Számos bizonytalanság van még tehát, azonban egészen elképesztő, hogy a mai élő összes élőlény közös őséről (a LUCA-ról, ami már körülbelül 4,5 milliárd éve is létezett) már tudjuk, hogy már rendelkezett CRISPR-CAS9-rendszerrel. Ez tulajdonképpen a baktériumok immunrendszere, melynek révén a baktérium a genomkönyvtára alapján az őt megtámadó vírust felismeri, és szétvágja annak DNS-ét. Ez a roppant fontos eljárás például a génsebészet és számos már eljárás, kutatás alapja. Többek között ahhoz is hozzájárulhat, hogy két szülő genomjának ismeretében megakadályozzák a születendő csecsemőbe való káros génszekvenciák átörökítését, megelőzve így például halálos kórokat. Természetesen számos morális és etikai kérdés felmerülhet a hasonló eljárásokkal kapcsolatban, arról nem is beszélve, hogy ilyenfajta mesterséges szelekcióra még nem volt példa. De kétségkívül ez már nem a jövő, hanem a mindennapjaink. Egy biztos: a genomika haszna az orvoslásban az egyénre szabott medicina felé fogja lendíteni az egészségügyet, és a szekvenálás, a genetikai feltérképezés egyre olcsóbb, így egyre szélesebb körben elérhetővé válik, ez pedig erősíti a preventív diagnosztikát. Hiszen ha előre tudom, hogy egy betegségre magas a rizikófaktor, akkor tudatos életmóddal elkerülhető annak kialakulása, súlyossá, krónikussá válása.
Ember, vigyázó szemed a genomikára vesd!
Felhasznált források:
https://regi.tankonyvtar.hu/hu/tartalom/tamop425/0019_1A_Nepegeszsegugyi_genomika/ch02.html